Evolution ‘Landscapes’ Predict What’s Next for COVID Virus
Mathematically modeled “landscapes” that describe the relative fitness of various mutations can in theory be used to project how SARS-CoV-2 and other viruses could evolve in the future.
Olena Shmahalo for Quanta Magazine
Introduction
In the fall of 2019, the world began one of the largest evolutionary biology experiments in modern history. Somewhere near the city of Wuhan in eastern China, a coronavirus acquired the ability to live inside humans rather than the bats and other mammals that had been its hosts. It adapted further to become efficient at spreading from one person to the next, even before the body’s defenses could rise against it. But the evolutionary chess game didn’t stop there, and we have a Greek alphabet soup of SARS-CoV-2 variants to prove it.
Researchers around the world are trying to understand the virus’s evolution in more detail, and particularly how mutations in SARS-CoV-2 alter its ability to spread among humans. “A well-adapted virus today could be maladaptive tomorrow as the host develops resistance, and then it has to figure out a new way to infect that host. That drives the innovation that drives the novelty,” said Justin Meyer, an evolutionary biologist at the University of California, San Diego.
Grim as the human toll from the constantly shifting pandemic is, the abundance of scientific data from watching the virus evolve as it moves around the globe has been instructive. “COVID has given us some of the most beautiful examples of evolution in action,” said Luca Ferretti, a statistical geneticist at the Big Data Institute of the University of Oxford.
Predicting exactly what the virus may do next may never be possible, but virologists around the world have been gaining insights into which components of SARS-CoV-2 are most prone to evolve and which key protein elements can’t change without tanking its survival. That information could point the way to better, more enduring vaccines. Other studies have highlighted ways in which the virus could evolve resistance to the monoclonal antibody therapies used to treat some severely ill COVID-19 patients. The work has also pinpointed specific combinations of mutations that, if they become widespread in the viral population, could usher in a new phase of the pandemic driven by variants that excel at evading our immune defenses in addition to spreading quickly.
Scientists have been able to make these discoveries by revisiting a concept proposed almost a century ago — fitness (or adaptive) landscapes — with modern technologies. They can use fitness landscapes to quantify the relationship between changes to the viral genome and its ability to replicate and infect a new host. The topographic maps representing that relationship can help to reconstruct the virus’s history, and they could also at least potentially predict its future.
To Tobias Warnecke, a molecular evolutionary biologist at Imperial College London, fitness landscapes are an invaluable way to connect genotype to phenotype. By tapping into their quantitative potential, he says, scientists can ask questions about how two mutations affect a trait in concert, and how they might be influenced by the presence of a third mutation. “In that way,” he said, “you can go through many different combinations of genotypes and see how that affects whatever you’re interested in.”
The value of fitness landscapes isn’t limited to comparisons between small numbers of changes in genomes or proteins. Modern experimental techniques enable a strategy called deep mutational scanning, in which researchers perform a small-scale experiment in natural selection and compare the fitness value of tens of thousands of mutant variants at once. The process can reveal unforeseen interactions between mutations that can help or hurt a virus — and it can identify paths for the future evolution of a virus that might pose new threats to humans.
A Dynamic Map for Survival
In On the Origin of Species, Charles Darwin wrote that natural selection was the result of the “preservation of favorable individual differences and variations, and the destruction of those which are injurious.” In those days, before the scientific understanding of genetics and mutations, biologists could only try to imagine how small, inheritable changes to an organism could impact its reproduction. The idea fully solidified only with work by the American biologist Sewall Wright. In his seminal 1932 paper in the Proceedings of the Sixth International Congress of Genetics, he used hand-drawn diagrams to illustrate how an organism might move through the “almost infinite field of possible variations through which the species may work its way under natural selection.”
Wright noted that one way to visualize the vast number of possible variants of linear molecules like DNA or peptides was to treat each possibility as a unique point in space. Evolution of the molecule then equates to a path between the points for the initial and final variants that hits all the points for intermediate variants along the way.
As an aid to understanding the complex graphs of these variants and the evolutionary paths between them, Wright showed that they can be represented as more intuitive “adaptive landscapes” of just two or three dimensions. The horizontal axes plot the variability in DNA (genotypes) or physical traits (phenotypes); the more similar two variants are, the closer they sit on the plane. The vertical axis measures the impact of the variation on evolutionary fitness. Variants that improve an organism’s odds of surviving, whether by increasing its viable offspring or improving the function of its proteins, perch on peaks, while those that diminish it languish in valleys.
What results is a landscape with a unique topography, explains Adam Lauring, an evolutionary biologist at the University of Michigan Medical School. If the mapped variants don’t differ much in their impact on fitness, then the landscape looks fairly flat, much like Nebraska. Variants with large effects on fitness create a landscape that more closely resembles the towering hoodoos of Bryce Canyon in Utah. Natural selection favors the variants on peaks: The average genotype or phenotype of a species should evolve by moving from one peak to the next, ideally along a ridge between them rather than through the valleys. (Isolated subpopulations with different genotypes can also help a species find its way over a gap.)
“If you move a few feet, you’re going to fall off, and getting up again is getting very hard,” Lauring said. “There are fewer pathways to move around.”
“The theory is very straightforward. You just need to know your genotype, and then you measure the fitness and you can basically predict anything that might happen,” said Claudia Bank, who researches evolutionary dynamics at the University of Bern in Switzerland. But putting the theory into practice is another matter.
One complication is that a fitness landscape, whether for SARS-CoV-2 or a human, isn’t static. A mutation that lets an organism digest a new food but makes it grow more slowly could be either a lifesaver or a lethal handicap. A variant’s impact on evolutionary fitness depends on the environment in which an organism lives. When the environment changes, so does the fitness landscape. “Different mutations have different impacts, and that depends on the fitness landscape,” Lauring said.
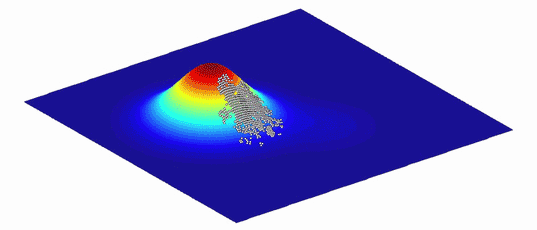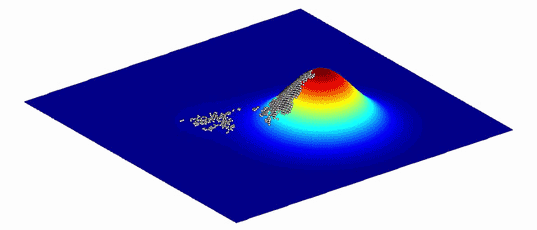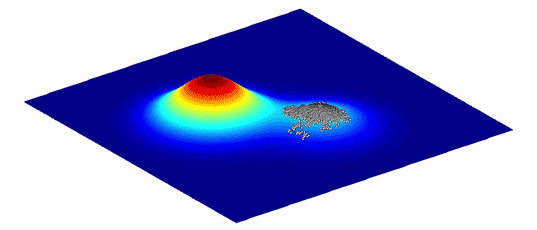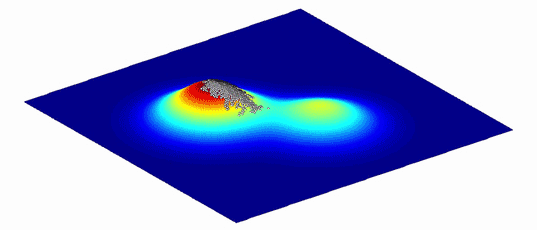
Because environments change constantly, the fitness landscapes are also always in flux. Populations of organisms evolve to follow the peaks.
Creating fitness landscapes is also a mathematical challenge. Even a small protein just 100 amino acids in length will have 20100 possible variants, more than the number of atoms in the universe. It’s hard to imagine, let alone compute, the complex topographies of fitness landscapes for real proteins and the likelihood of various paths across them. Consequently, for decades fitness landscapes were conceptual aids rather than tools for concrete measurements. Only recently, with advanced computing power and improved molecular biological technology, have scientists been able to start making quantitative landscapes for individual proteins and simple organisms like bacteria and viruses.
Bacteria and viruses are almost ideal subjects for fitness landscapes. Growing by the millions or billions in a test tube, each bacterial cell or viral particle can harbor one mutation from the huge pool of variants that describe the fitness landscape. Their short generation times, on the scale of hours or days, also allow researchers to identify new mutations much more quickly. Most viruses that use RNA as their genetic material, including HIV and the hepatitis C virus (HCV), are also highly prone to mutation because the RNA polymerase that replicates their genome doesn’t proofread the copies as effectively as DNA polymerases do.
One of the first things scientists began to discover is that despite the complexity of the landscapes, organisms are often constrained to just a handful of fitness maxima and a limited number of pathways between them. A 2006 Science paper took a close look at a protein called beta-lactamase, which inactivates antibiotics such as penicillin. The joint effects of five single-nucleotide mutations in beta-lactamase can increase its antibiotic resistance by a factor of 100,000. With his colleagues, Daniel Weinreich, an evolutionary biology postdoctoral fellow at Harvard University at the time who now heads a laboratory at Brown University, noted that the evolution of the gene could potentially follow 120 paths to accumulate all five mutations.


Samuel Velasco/Quanta Magazine; source: doi.org/10.1126/science.1123539
However, when the scientists created and tested the intermediary variants in the lab, they found that 102 of the paths weren’t possible under natural selection because they produced defective or incomplete proteins. The possibilities narrowed further when they found that many of the remaining combinations failed to improve antibiotic resistance. “This implies,” they wrote, “that the protein tape of life may be largely reproducible and even predictable.”
Deep Mutational Scanning
But predicting the future evolutionary trajectory of even the smallest virus or protein requires a detailed knowledge of its fitness landscape, which is hard to obtain. Historically, scientists had to create mutations one nucleotide or amino acid at a time, then purify the mutant protein and assess its function. It was often impractical to examine more than a few of the possible mutations.
The development of technologies for deep mutational scanning changed all that. This technique allows scientists to generate tens of thousands of variants in one go, and then make all the variants compete against one another to determine their relative fitness value.
Researchers start by creating a library of variant genes that can be cloned into cultured cells. The genes code for a protein whose activity is linked to some biochemical function that can be selected for in the laboratory, so the cells making the “fittest,” most active versions of these proteins will become more abundant, while cells making inactive versions disappear. With high-throughput DNA sequencing, researchers can then tally up the numbers of each variant for a quantitative measurement of how well it performed over multiple generations.
“It’s a really powerful approach to capture the impact of mutations,” said Valerie Soo, a researcher in Warnecke’s laboratory in London.
With mutation-prone RNA viruses, scientists don’t even have to generate variants in the lab — the error-prone genomic replication machinery introduces mutations and does the job for them. Each of the millions of copies of the virus is slightly different from its neighbors, creating what virologists call a mutant swarm. Within this swarm is the raw material of evolution by natural selection.
“Microbes reproduce so rapidly that evolution happens on a daily basis. You can actually monitor evolution in real time,” said Samuel Alizon, an evolutionary ecologist at the MIVEGEC laboratory in Montpellier, France.
Researchers found that very few of the mutations in those swarms get passed on to new hosts, particularly when only a small amount of virus is required to cause an infection. Some of this is pure chance, a matter of which variant is in the right place at the right time. But by sketching out fitness landscapes, researchers can try to figure out why some variants are transmitted far more frequently than others, says Raul Andino-Pavlovsky, a virologist at the University of California, San Francisco.
“A virus not only needs to be able to generate diversity, but it has to be able to tolerate this diversity,” he said. “If you’re a virus and you can tolerate changes, you’re likely to be a virus that has much better capacity for adaptation.”


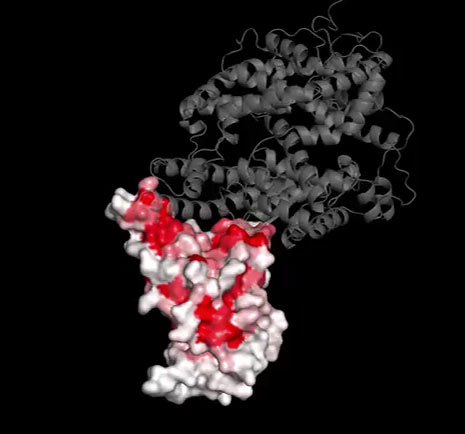
The researchers Tyler Starr (top) and Jesse Bloom of the Fred Hutchinson Cancer Research Center have studied a crucial domain of the SARS-CoV-2 spike protein to find out which parts of it tend to draw the attention of the immune system.
The researchers Tyler Starr (left) and Jesse Bloom of the Fred Hutchinson Cancer Research Center have studied a crucial domain of the SARS-CoV-2 spike protein to find out which parts of it tend to draw the attention of the immune system.
Courtesy of Tyler Starr; Courtesy of Jesse Bloom
Fitness landscapes are the perfect way to describe, both conceptually and quantitatively, how viruses from chronic or persistent infections evade repeated efforts to neutralize them by their host’s immune system, according to the evolutionary biologist Tyler Starr. It’s why he joined the lab of Jesse Bloom at the Fred Hutchinson Cancer Research Center to study how HIV coevolves with antibody immunity inside a patient over the course of an infection. His goal was to understand how this evolutionary arms race between a virus and the immune system yields antibodies with protective properties, which could help scientists developing an HIV vaccine to focus on the more immutable parts of the virus.
But no sooner had Starr begun his work on HIV than another virus stole his — and the world’s — attention.
More Mutable Than Expected
As SARS-CoV-2 began its global spread, Starr and Bloom realized that fitness landscapes provided a useful way to begin studying the novel pathogen. It gave them a way to figure out what factors were important in viral proteins and how much change the virus could tolerate.
Initially, scientists sequencing SARS-CoV-2 didn’t notice much genetic variation. Although coronaviruses use an error-prone RNA polymerase to copy their genetic material, SARS-CoV-2 has a second protein that acts as a proofreader. So researchers didn’t expect the virus to acquire as many mutations as, say, influenza or HIV.
Bloom and Starr knew that the spike protein would be the part of the coronavirus under the most intense evolutionary pressure because it is what the immune system recognizes most strongly and what the virus uses to break into the body’s cells. With 1,273 amino acids, however, the spike protein is too sizable for rapid evaluation by a fitness landscape. Starr therefore decided to focus on a subsection of the spike protein known as the receptor binding domain, which is just a few hundred amino acids — a much more tractable problem.
Starr used deep mutational scanning to create 4,000 different mutations of the receptor binding domain. He evaluated their ability to bind to the human ACE2 protein (the molecular “lock” it picks to enter cells) and to be recognized by the immune system. If SARS-CoV-2 couldn’t tolerate much variation in its receptor binding domain, Starr expected to see that the immune recognition or ACE2-binding functions would be severely compromised by mutations.
But that’s not at all what happened. “The receptor binding domain had a lot of different mutations that actually improved binding affinity,” Starr said. “This looked like a really tolerant domain that had a lot of capacity to evolve. Yet the mindset at the time was that coronaviruses don’t evolve antigenically. They were probably going to be stable.”
While the receptor binding domain tolerated more variation than expected, not all parts of the spike protein did. These parts of the spike protein may therefore be good targets for new vaccines and monoclonal antibodies, Starr says, since they are less likely to mutate over time.
When they first posted these results on the biorxiv.org preprint server in June 2020, it was a huge wake-up call, Starr says — one of the first indications that SARS-CoV-2 was more mutable than people thought. Now Starr and Bloom are repeating their deep mutational scanning experiments on the alpha, beta, gamma, delta and omicron variants to gain similar insights about their receptor binding domains.
Starr, Bloom and colleagues also created a map of all the possible mutations to the receptor binding domain that didn’t interfere with ACE2 binding. Their work, published in Science in January 2021, identified potential mutations in this domain that could evade neutralization by monoclonal antibody therapies. Their work also identified several mutations that emerged in an immunocompromised individual who was infected with SARS-CoV-2 for 150 days. By the time this person received monoclonal antibody treatment at day 145, they had already developed resistance to the available products on the market. To Starr, this showed that these therapeutic monoclonal antibodies could become less effective over time, either within a single patient or more generally as the virus mutates.
Moreover, as Starr, Bloom and their colleagues described last summer in Nature Communications, several widespread mutations can each help SARS-CoV-2 evade some of the antibodies that the immune system typically directs against the most targeted parts of the receptor binding domain. So far, no viral lineage has evolved to have all three of these mutations. “However, we suggest the appearance of such a variant would be a worrying development and should be monitored closely,” they wrote.
The world in which SARS-CoV-2 first emerged at the end of 2019 was different from the world of today. The ability of the virus to produce lots of copies of itself and to spread between individuals was surely key to its success early in the pandemic. As the number of people immunized through vaccination and naturally acquired infection rises, however, the virus will experience more pressure to evade immune responses. Lauring says many mutations come with trade-offs, and SARS-CoV-2 is no exception. An immune escape variant with reduced virus transmission might not have been favored in early 2020, but it might be now.
“We’re the environment for the virus,” Lauring said. “If we change, the landscape changes.”



{kind=link}