Genetic Architects Untwist DNA’s Turns

Juj Winn for Quanta Magazine
Introduction
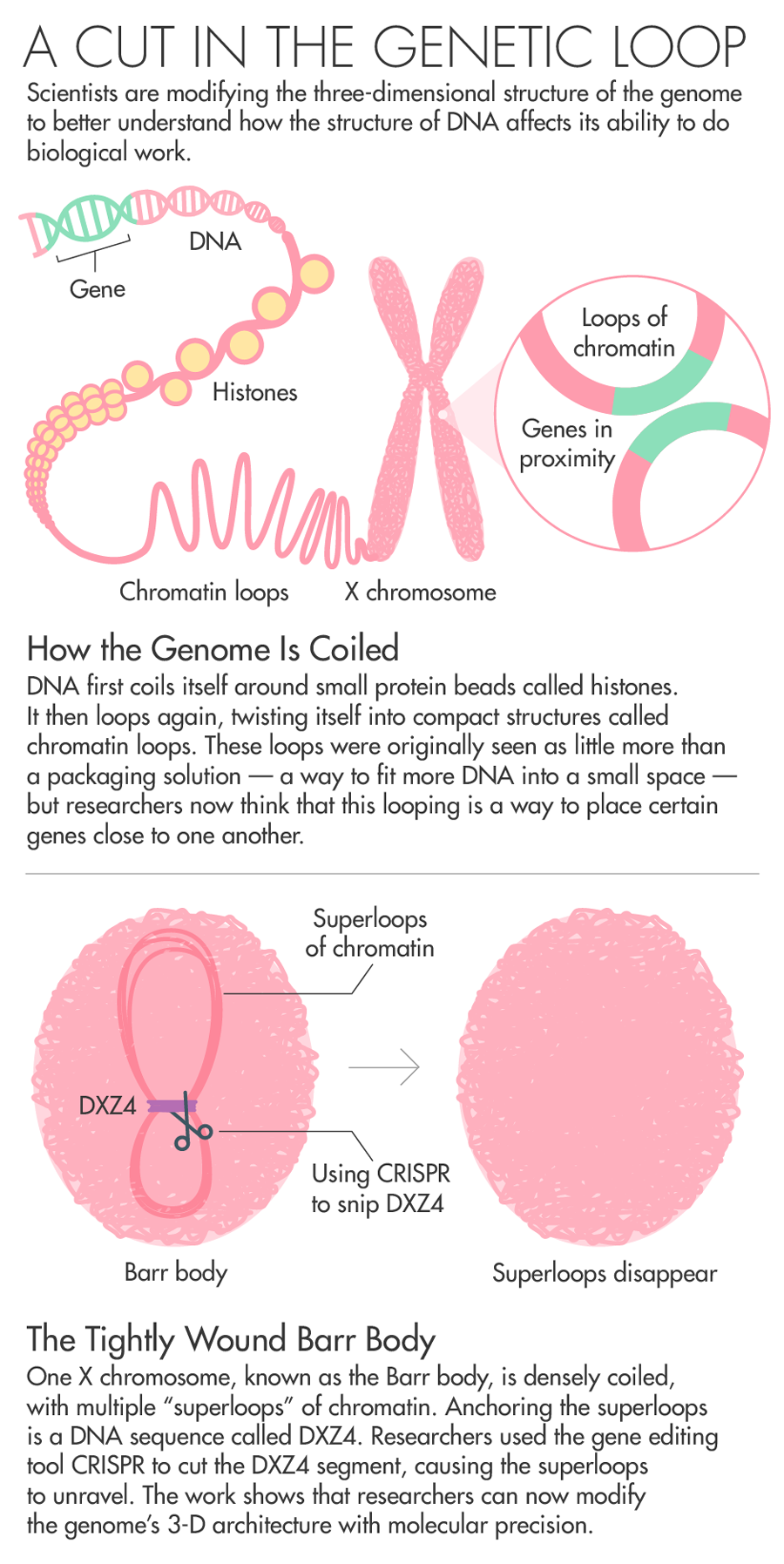
When scientists finished sequencing the human genome in 2003, many researchers focused their attention on decoding the long strings of As, Ts, Gs and Cs as the way to understanding the mysteries of human genetics. The genome, however, doesn’t appear in nature as a simple long line of letters. Unfurled, the genome stretches for nearly two meters, yet it folds itself in coils and loops to fit inside a nucleus less than 10 microns in diameter.
In recent years, researchers have begun to understand just how important this 3-D genetic architecture really is. Just as the only accessible information in a book is what’s on the open page in front of you, the genome’s instructions can only be read by the cell when those instructions aren’t hidden inside deep folds. But researchers don’t understand how the cell folds DNA in such a way that the important bits can be read.
Earlier this year, two independent groups of researchers took a big step toward decoding the mysteries of DNA folding. Both groups used the powerful new gene editing tool known as CRISPR to uncoil a tightly wound piece of DNA. Their work could help scientists identify some of the basic rules behind how and why the genome forms its 3-D structure. It also reveals that the presence of small DNA sequences can lead to massive changes in how the genome is organized.
The new research began as an attempt to understand a mystery of the X chromosome. Most male mammals have an X and a Y chromosome, while most females have two Xs. This presents a potential problem for a female’s cellular machinery. If both Xs remain active, twice the number of X chromosome genes will be switched on. This will lead to a host of developmental difficulties, often culminating in the death of the embryo soon after fertilization.
To avoid this scenario, one copy of the X chromosome switches itself off using a gene called Xist. The inactive X chromosome, known as the Barr body, is small. It coils itself up and hides nearly all of its genes from the cellular machinery that turns DNA into RNA and proteins.
“X inactivation is a critical process in female development,” said Brian Chadwick, a biologist at Florida State University. “The only difference between the two Xs is how they’re packaged.”
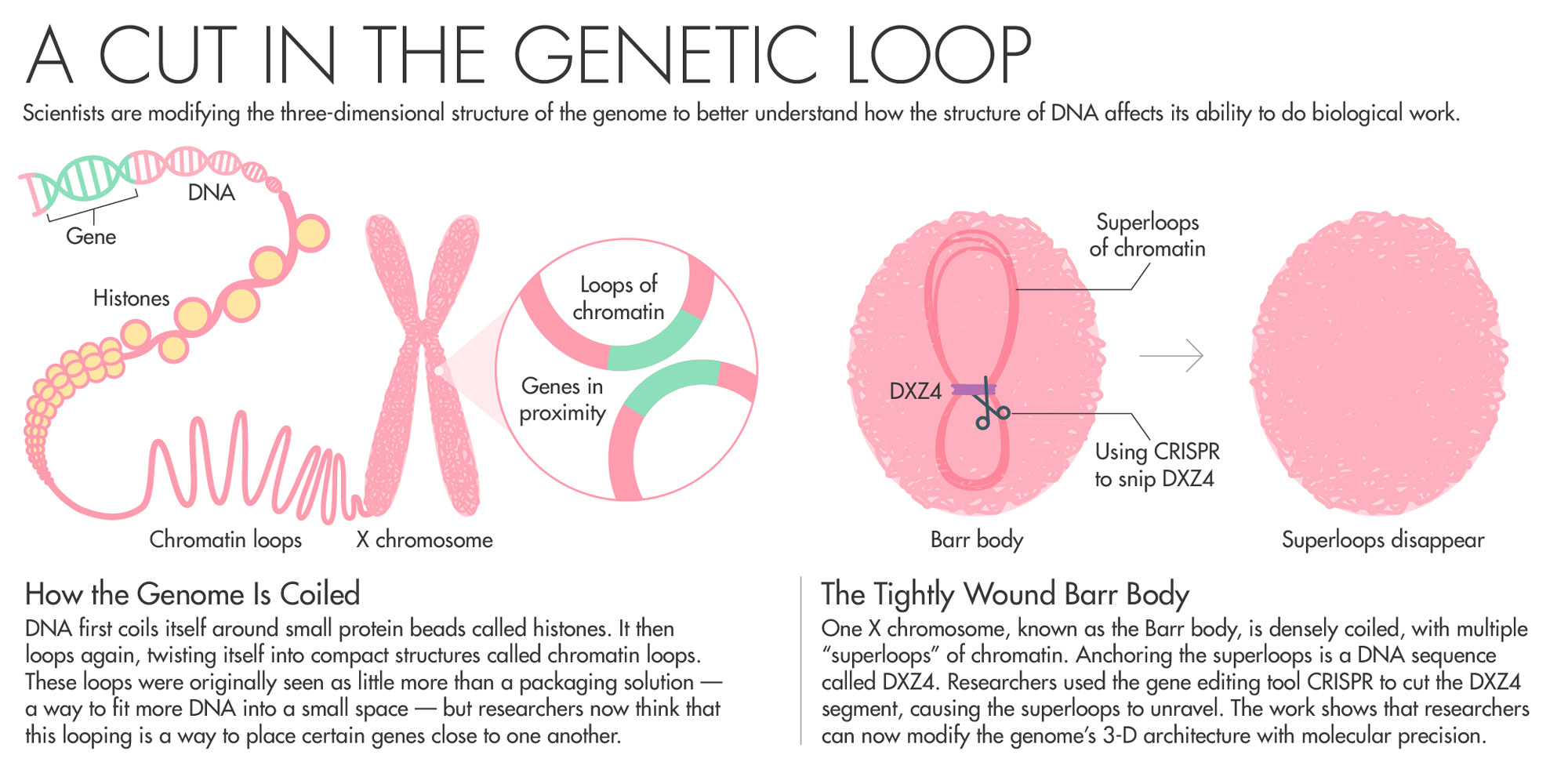
Chadwick has spent his entire career trying to understand how the body inactivates one copy of the X chromosome. Chadwick’s work on Barr bodies zeroed in on a DNA sequence called DXZ4 as being potentially key to the folding that switches off most of the genes on one copy of the X chromosome. All the mammals his team has looked at with an inactive X chromosome had these sequences.
But how was Chadwick to test whether DXZ4 really was the origami master of the X chromosome? A major clue appeared in 2014, in work that had begun over two decades before.
Lucy Reading-Ikkanda for Quanta Magazine
The First Connections
As a doctoral student in Mark Seyfred’s lab at Vanderbilt University in the early 1990s, Katherine Cullen wanted to understand a protein called prolactin, which enables female mammals to make milk. Cullen and Seyfred believed that exposure to estrogen sets off a cascade of events that ends in the creation of a giant loop of DNA. This loop connects the prolactin gene promoter, which acts as a switch to turn the gene on, with a prolactin enhancer, which serves as the metaphorical finger that flips the switch. Cullen’s task for her doctorate was to verify this.
She searched for this loop using a technique that had recently been developed by Seyfred called a proximity ligation assay. The first step is to treat the genome with formaldehyde, which creates cross-links between segments of DNA that are close to one another in the genome’s natural 3-D form. The process reveals which parts of the genome are touching even if those sequences are far away from one another on the linear genome. Cullen found that exposure to estrogen creates a loop that links the prolactin enhancer and the promoter. Her discovery earned her a 1993 publication in Science and sent her well on her way to her doctorate.
Cullen’s work offered some of the first direct evidence that the larger three-dimensional structure of the genome is related to its function. But her article didn’t receive much attention. “I wasn’t thinking about genome architecture at the time. The term wasn’t even a term back then,” Cullen said.
Things had changed a decade later, when she received a surprise call from Erez Lieberman Aiden, at the time a visiting fellow at Harvard University, who was looking for more information about the ligation assay she had used. Aiden had big ambitions for it. He didn’t want to just use the assay on one gene. Instead, he wanted to search through the whole genome, identifying not just one loop but potentially thousands of them.
Over the next few years, Aiden, who is now a computational biologist at Rice University and Baylor College of Medicine, built on work by Job Dekker, a biologist at the University of Massachusetts Medical School, to create the Hi-C system, which maps the probability that two pieces of the genome are touching each other.
“We bump into our next-door neighbor more than someone who lives in another country. It’s the same idea here,” Aiden said. “If we know who bumps into whom in an ordinary day of the genome, then we can figure out how close these areas are and consequently what the genome looks like in three dimensions.”
Aiden and colleagues published results of the initial Hi-C experiments in 2009. These results revealed genome architecture at 1-million-base-pair resolution — a pointillist outline that began to reveal the genome’s dazzling complexity. “The results were like a map of the world that only gave you the fuzzy outlines of the continents,” said Suhas Rao, a graduate student who works in Aiden’s lab. “You couldn’t really navigate with them, but it was a place to start.”
The Aiden lab spent the next few years refining Hi-C and, in 2014, published a paper in Cell that charted every loop and coil of DNA at 1,000-base-pair resolution. If the 2009 paper revealed the blurry outlines of North America, the new Hi-C exposed a street grid of Manhattan. The detail gave scientists some of their first clues about the rules by which the genome folds.
Of all the billions of contacts among DNA that Aiden’s Hi-C generated, one area of the genome stood out. In XX female cells, Rao, Aiden and colleagues found that while one X shared patterns of loops with the rest of the chromosomes, the inactive X looked very different. Instead of having multiple loops of around 200,000 base pairs, the inactive X had two massive “superdomains” characterized by multiple “superloops” of up to 77 million base pairs. What was anchoring the superloops? A DNA sequence called DXZ4 — the same one that Chadwick had previously identified as key to the folding of the X chromosome. Chadwick read the paper and reached out to Aiden. The pair agreed to collaborate.
A Genetic Cut
Understanding the relationship between the structure of a molecule and its function is a classic question in biochemistry. Scientists studying proteins mastered this field of inquiry beginning in the 1960s by changing one amino acid building block in a protein and measuring how it altered the protein’s ability to do its job. Chadwick and Aiden wanted to do something similar to understand the relationship between DNA sequences and genome architecture. Like many genetics labs, they turned to the genome editing tool CRISPR, which can act as a set of biomolecular scissors.
To prove that DXZ4 really does influence genome folding, the team took human cells and used CRISPR to snip out the DXZ4 section — a repetitive stretch of DNA that goes on for hundreds of thousands of nucleotides. They then used Hi-C to measure how the cut affected chromosome looping. When they removed DXZ4, “those gargantuan loops disappeared. The chromosome starts to look like a normal autosome,” Aiden said. “It showed that we could have fine control over how the genome folded.”
Independently, Dekker’s lab had likewise shown the key role of DXZ4 in the folding of the inactive X chromosome in mice. They also revealed that the Xist gene — the molecular switch that coils up the inactive X chromosome — helps to create the boundary between the two large superdomains on that chromosome. Both Dekker’s paper (published in Nature) and Aiden and Chadwick’s (published in the Proceedings of the National Academy of Sciences) have helped to untangle the Gordian knot of genome folding.
“It’s quite spectacular that the structure of an entire chromosome would rely on a small DNA sequence somewhere in the middle,” said Wouter de Laat, a biomedical geneticist at Utrecht University in the Netherlands.
These papers, de Laat said, are expanding our knowledge of the intimate relationship between how a genome folds and how it functions. Scientists have long suspected that abnormal genome folding may cause diseases, and several new studies have identified links between genome architecture and biological development. A 2016 study by Stefan Mundlos at the Max Planck Institute for Molecular Genetics in Berlin and his colleagues showed that a rearrangement of DNA in a noncoding region of the genome caused limb malformations during development by changing chromatin folding. Other researchers are using CRISPR to investigate whether changes to genome architecture affects the ability of parasites like Trypanosoma, the cause of African sleeping sickness, to evade the immune system.
It’s becoming apparent that in the genome, as Dekker says, “nothing makes sense except in 3-D.”
This article was reprinted on Wired.com.



